

Differential of paroxysmal events
Paroxysmal dyskinesias are characterized by recurrent episodes of sudden involuntary movement disorders.
These disorders are commonly associated with other episodic symptoms, such as migraine and episodic ataxia. Although paroxysmal dyskinesias were thought to be channelopathies (and some are), none of the three genes which are associated with the bulk of these conditions actually encode ion channels.
Classification
Paroxysmal dyskinesias are classified according to the precipitating factors into three major subtypes:
- Paroxysmal Kinesigenic Dyskinesia (PKD)
- Paroxysmal Nonkinesigenic Dyskinesia (PNKD)
- Paroxysmal Exercise-Induced Dyskinesia (PED)
In addition, a further paroxysmal entity occurs, Paroxysmal Hypnogenic Dyskinesias (PHD) characterised by attacks occurring during sleep without identifiable trigger), and usually due to the condition of autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) in most cases1.
Clinical Features
All three forms can present with dystonic, choreic, or ballistic movements, or a mixture of those, and can involve one or more limbs, the trunk, and/or the face.
1. PKD (Paroxysmal Kinesigenic Dyskinesia)
PKD is the commonest type, and dystonia the commonest movement disorder.
Virtually all cases due to mutations in the PRRT2 gene have a clear kinesigenic trigger, although in up to 40%–50% of cases anxiety, stress, startle or prolonged exercise can also induce attacks2.
Very rarely (about 1%–2% of patients) there are no kinesigenic triggers. About half of the patients experience a sensory aura at the initial site of the attacks.
Genetics
PRRT2 mutations are common in patients with PKD and are significantly associated with an earlier age at onset and longer duration of attacks3. PRRT2 mutations account for approximately 60% to 100% of patients with familial PKD and 10% to 50% of sporadic PKD cases. Inheritance follows an autosomal dominant pattern. In patients with homozygous or compound heterozygous mutations there is a more severe phenotype.MCG
About 25% of patients with PRRT2 related PKD have infantile convulsions4. In patients with Infantile Convulsions with Choreoathetosis (ICCA) syndrome, PKD starts after the onset of epilepsy (the seizures develop within the first two years of life), usually after age five, although some patients might have epileptic seizures at a later age.
After a peak in puberty, there is a tendency for the attack frequency to decrease, and the condition may completely remit in adulthood4.
Mutations in the PRRT2 gene are also associated with Benign Familial Infantile Seizures (BFIE) (brief seizures with cyanosis, increased tone and jerks of the limbs, which remit by two years of age). PKD, BFIE and ICCA are considered to represent a continuous disease spectrum.MCG
| Trigger | Aura | Duration | Frequency | Movement | Pain | Age at Onset | |
| 1.PKD |
Sudden movement/ startle ; modification of ongoing movement; anxiety, stress, or prolonged exercise.
Can be self-provoked. |
Frequently present | Brief Usually lasting < 1 minute and most commonly less than 10 seconds |
20 attacks per day (1/month to 100/day)
80% of patients reported tens to hundreds of attacks per day4 |
Both chorea and dystonia, and tend to generalise. Ballism may be present. |
No | Childhood, very rarely later than 20 years of age |
| 2.PNKD |
Coffe, alcohol and stress. Anxiety, excitement. |
May be present |
Long
10 minutes to 4 hours |
Infrequent, may be weekly4 or a few times a year.
(Rarely > 1 daily). May be attack free for months. |
Both chorea and dystonia. | Possible | Childhood |
| 3.PED | Exercise Hyperventilation |
Intermediate: 5-30 minutes |
Weekly | Both chorea and dystonia. May involve body part involved in exercise; legs typically involved. | No | Childhood-30 years |
This patient was a 20â€yearâ€old man who had experienced 2 attacks of infantile convulsions at the age of 5 to 10 months and had involuntary dystonia, usually alternating between the left and right sides, since the age of 10. He would experience increased muscular tension in the lower limb of the affected side before an episode, and slowing down occasionally relieved the attack. The episodic attacks, mostly triggered by voluntary movement and emotional stress, but never by caffeine, occurred 10 to 30 times per day and usually involved the face and neck. Each attack lasted less than 20 seconds and was never associated with loss of consciousness.
(vv)PatientR.mp4(tt)
From: Tian WT, Huang XJ, Mao X, Liu Q, Liu XL, Zeng S, Guo XN, Shen JY, Xu YQ, Tang HD, Yin XM, Zhang M, Tang WG, Liu XR, Tang BS, Chen SD, Cao L. Proline-rich transmembrane protein 2-negative paroxysmal kinesigenic dyskinesia: Clinical and genetic analyses of 163 patients. Mov Disord. 2018 Mar;33(3):459-467.
(vv)Slc-3.mp4(tt)
Patient RJ7712 was a 19â€yearâ€old woman who had involuntary dystonia affecting the right or both sides since the age of 15. The episodic attacks, mostly elicited by voluntary movements, occurred 10 to 20 times per day, with an approximate duration of 10 seconds Carbamazepine (50 mg/night) could satisfactorily control the attacks. She experienced an incomplete remission at the age of 19, with occasional aura attacks
From: Tian WT, Huang XJ, Mao X, Liu Q, Liu XL, Zeng S, Guo XN, Shen JY, Xu YQ, Tang HD, Yin XM, Zhang M, Tang WG, Liu XR, Tang BS, Chen SD, Cao L. Proline-rich transmembrane protein 2-negative paroxysmal kinesigenic dyskinesia: Clinical and genetic analyses of 163 patients. Mov Disord. 2018 Mar;33(3):459-467.
2. PNKD
There is always a dominant family history for similar attacks, no sporadic cases having been reported thus far. Attacks are generalised in about 50% of the patients.
In a number of patients episodes became heterogeneous during the course of the disease, being more dystonic in the early phase, with more choreic components seen subsequently4.
Attacks can rarely be complicated by dysarthria, dysphagia, oculogyric crises, inability to move and may even be fatal.
The duration of the attacks is variable but typically last from several minutest to 1-2 hours.
Although several non-kinesigenic triggers (ie, stress, tiredness, sustained exercise) can be present, the attacks are characteristically brought on by coffee and/or alcohol intake.
3. PED
The majority of patients showed focal/unilateral involvement, and generalization of the attack is uncommon4. lower-limb dystonia precipitated by sustained walking or running is the most common manifestation. Most cases with PED are de novo, whereas only 10% have a positive family history.
Associated neurological disorders, seen in almost ¾ of patients include epilepsy, learning difficulties, ataxia, and pyramidal signs4.
Genetics
A mutation in the solute carrier family 2, member 1 (SLC2A1) gene is the most common cause of PED. SLC2A1 encodes GLUT-1 which is the primary glucose transporter into erythrocytes, across the blood–brain barrier and into and out of astrocytes. Patients usually show CSF glucose and lactate levels below the 10th percentile. Lactate levels are typically also low, a CSF to blood glucose ratio at or below the 25th percentile, and CSF lactate levels are never elevated5. (Clincially, these patients have epilepsy, hypotonia, spasticity, ataxia, and developmental delay2).
Segment 1. Recording of a running session. This segment shows involuntary bilateral hand twisting movements (white arrows) presenting at the end of the sprint of a short running path. The patient complains also facial grimace and leg tightness (not shown).
Segment 2.Neurological examination and provoking maneuver (running in place). This segment shows an unremarkable finger-tapping test in the beginning of the examination. The exercise provokes after 10 seconds the onset of bilateral hand dystonia with left clenched fist and right hand twisting posture. The finger tapping performances at the end of the segment are impaired by bradykinesia and dystonic jerks
(vv)Vid1PED.mp4(tt)
From: Marano M, Motolese F, Consoli F, De Luca A, Di Lazzaro V. Paroxysmal Dyskinesias in a PRRT2 Mutation Carrier. Tremor Other Hyperkinet Mov (N Y). 2018 Dec 3;8:616. doi: 10.7916/D8S488X0. PMID: 30622840; PMCID: PMC6315045.
Major differential includes Dopa-responsive dystonia and mutations in the parkin gene. Note that currently a gene panel is likely to be most cost effective method of making the diagnosis. Typically, this should be obtained before imaging.

From: Erro R, Stamelou M, Ganos C, et al. The Clinical Syndrome of Paroxysmal Exercise-Induced Dystonia: Diagnostic Outcomes and an Algorithm. Mov Disord Clin Pract. 2014;1(1):57-61. doi:10.1002/mdc3.12007
4. PHD
Characterized by violent attacks of dystonic and tonic movements that occur during sleep and last for around 45 sec. In fact, PHD is almost always a form of frontal lobe epilepsy ‘autosomal-dominant nocturnal frontal lobe epilepsy’ (ADNFLE). Mutations have been found in CHRNA4, CHRNA2 and CHRNB2, which code for acetylcholine receptor subunits.
However, PRRT2 mutations have been recently identified in 2 out of 11 patients (18.2%) with PHD6.
Examination: See Table 1.
Age of onset: See Table 1.
Diagnosis & Special Investigations
The critical features on diagnosis are:
- Nature of the trigger (see Table 1),
- Presence of additional interictal neurological signs2:
Epilepsy |
Movement Disorders |
Migraine |
|
| 1.PKD/PRRT2 mutations |
|
Cerebellar Ataxia Also: PNKD and PED7 |
|
| 2.PNKD/ MR-1 mutations |
|
Migraine
|
|
| 3.PED/SLC2A1 | Epilepsy | Ataxia |
Migraine |
Note: Low frequency of PRRT2 mutations in Episodic Ataxia and Hemiplegic Migraine

*If SLC2A1 is negative, also consider PRRT-2, especially if PNKD attacks are brief.
**Only 20-25% of PED cases are positive for SLC2A1, suggesting that other genes are implicated.
From: Erro R, Sheerin UM, Bhatia KP. Paroxysmal dyskinesias revisited: A review of 500 genetically proven cases and a new classification. Mov Disord. 2014;29(9):1108-1116. doi:10.1002/mds.25933
Prognosis
PKD: The frequency of attacks usually decreases with advancing age after puberty and the syndrome can completely remit regardless of any treatments.
Differential Diagnosis
There are a wide range of disorders which are reported to produce paroxysmal movement disorders (see Table 2)2,7.
The classical neurological examples include:
- Tonic spasms of multiple sclerosis and NMO ("brainstem fits")
- Episodic ataxias: eight EA syndromes are described according to their associated genetic loci. EA1 or EA2 are the most common disorders which are attributable to mutations in KCNA1 and CACNA1A respectively. Other EAs are rare and have only been reported in isolated case reports. Other identified causative genes include mutations in SLC1A3, CACNB4, and UBR4.
- Facio-brachio dystonia due to LGi1 encephalitis.
- Episodic axial hyperextension due to Progressive encephalomyelitis with rigidity and myoclonus (PERM), associated with glycine receptor alpha 1 antibodies..
- Limb shaking TIA
In addition, propriospinal myoclonus is frequently paroxysmal8.
| Structural lesions of CNS: | CNS and systemic immune disorders: | Cerebrovascular disorders: | Neurodegenerative disorders: | CNS infections: | Systemic metabolic disorders: | Other disorders: |
|
|
|
|
|
|
|
Paroxysmal attacks were initially reported in multiple sclerosis, but have since been described in neuromyelitis optica121. These attacks are identical phenomenologically to PKD.
It remains a matter of speculation regarding the antomical origin of these events: the majority likely have brainstem involvement, but hemispheric and spinal origins likely also occur.
The attacks typically are stereotyped and have the following characteristics:
Short duration (seconds to minutes)
High frequency of attacks, up to 200/day.
Consciousness is unaffected and EEG is normal, suggesting a nonepileptic origin.
The location of the precipitating lesion remains unknown but clinical findings are compatible with a brain stem origin.
Good response to carbamazepine
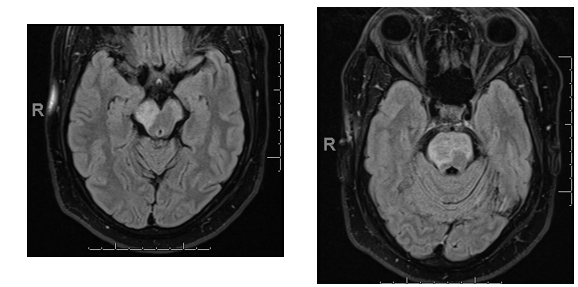
45 year old man, seen in the emergency room with recent onset of apparent seizures involving the left side of the body. History of hypertension and diabetes.
Stereotyped movements involving the left side of the body, including the face, and occurring currently throughout the day (20-30 times).
There was no associated loss of consciousness and no aura present. No history of epilepsy in the family. Not brought out, or worsened by movement. Resolved rapidly with phenytoin.
MRI show hyperintense signal in midbrain and pons on FLAIR images:
(vv)Pkd2.mp4(tt)
Similar events are described with infarction of the thalamus and of the midbrain (paroxysmal ataxia and dysarthria13):
The patient has an unusual hemdystonic gait, attempting to suppress involuntary movements of the right leg with her arm. Spontaneous dyskinesias of the right leg occur while sitting.
Finger-nose and heel-shintesting showed marked cerebellar dysmetria and tremor, but precise movements of the right-hand were normal. Elevation of the right leg resulted in ballistic jerks, with severe ataxia. The most remarkable feature of this presentation is the response to touch of the right arm and leg which led to complex movements with elements of jerks, dystonia, balance and stereotypies.
(vv)Tijssen.mp4(tt)
From: Nijssen PC, Tijssen CC. Stimulus-sensitive paroxysmal dyskinesias associated with a thalamic infarct. Mov Disord. 1992 Oct;7(4):364-6.
Functional paroxysmal dyskinesia:
Suggestive features include:
- Late age on onset; in one published series, the mean age at onset was 39 years, much later than that seen with other paroxysmal dyskinesias9.
- Paroxysmal Tremor may be present7, as may a range of other signs of functional neurological disorders.
- The movements are highly variable, and duration is typically much longer than paroxysmal dyskinesias, of the order of hours to days.
- Consciousness may be altered
- Attack triggers, although present, are not typical for paroxysmal dyskinesias; notably, 1 in 5 patients had coexistent organic movement disorder (tic, dystonia, tremor).
Treatment
| PKD |
Carbamazepine is the first-line treatment option, being very effective at low doses (50- 100 mg) (in 98% of patients3)(50–600 md/day). PRRT2-positive patients are more likely to respond11. |
| PNKD |
Clonazepam is the first-line pharmacological option when lifestyle modifications (i.e., avoiding coffee and alcohol) are not efficacious. Regardless of any treatment, there is a tendency for the attacks to reduce or remit in adulthood. Acetazolamide, valproate and levetiracetam may be of benefit. |
| PED |
PxD in the context of SLC2A1 cases have a positive but partial response to a ketogenic diet, which should be pursued to treat the underlying neuroglycopenia. Anti-epileptics not typically useful. |